
Resumo em linguagem simples
A Retinose Pigmentar afeta mais de 1,5 milhão de pessoas no mundo e é a distrofia retiniana hereditária mais comum. Entenda a genética, os sintomas, o diagnóstico e os tratamentos mais avançados — incluindo a terapia gênica aprovada e as novas abordagens gene-agnósticas em ensaios clínicos.
A doença que apaga a visão como um anel que se fecha
Começa com dificuldade para enxergar à noite. O paciente evita sair depois do anoitecer, tropeça em objetos que não estão diretamente à sua frente, demora mais para se adaptar quando sai de um ambiente iluminado para um escuro. Por anos, esses sintomas são atribuídos ao cansaço, à miopia ou simplesmente ignorados. Quando finalmente chega ao oftalmologista, o diagnóstico é revelado: Retinose Pigmentar.
A Retinose Pigmentar (RP) — também chamada de Retinite Pigmentosa — é a distrofia retiniana hereditária mais comum do mundo, afetando mais de 1,5 milhão de pessoas globalmente, com prevalência estimada em 1 caso para cada 4.000 habitantes. No Brasil, estima-se que entre 50.000 e 60.000 pessoas vivam com a doença, muitas sem diagnóstico formal.
Em 2025, a ciência avançou de forma significativa na compreensão e no tratamento da RP. Uma revisão publicada no Frontiers in Ophthalmology (Suleman et al., 2025) e dados do American Journal of Ophthalmology (Abbass et al., 2025) trazem perspectivas inéditas sobre epidemiologia, genética e terapias emergentes. Este artigo reúne o que há de mais atual, com a curadoria científica do Dr. Fernando Drudi e da Dra. Priscilla Almeida, especialistas em retina da Drudi e Almeida Oftalmologia.
O que é a Retinose Pigmentar?
A Retinose Pigmentar não é uma única doença — é um grupo heterogêneo de distrofias retinianas hereditárias que compartilham um mecanismo comum: a degeneração progressiva dos fotorreceptores da retina, com perda inicial dos bastonetes (responsáveis pela visão noturna e periférica) seguida pela degeneração secundária dos cones (responsáveis pela visão central e das cores).
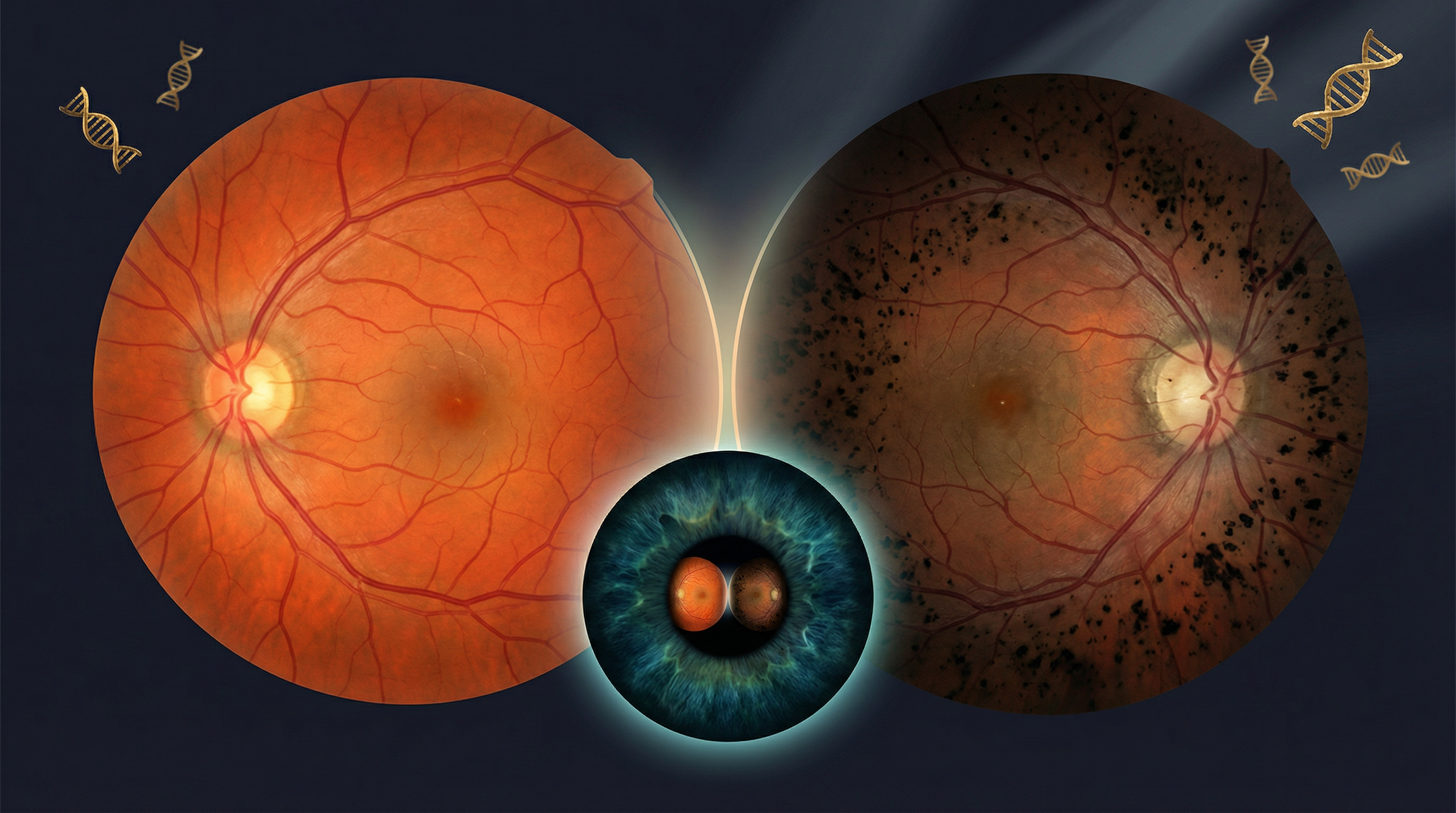
O nome vem do aspecto fundoscópico característico: depósitos de pigmento em forma de espícula óssea na retina periférica, acompanhados de atenuação dos vasos retinianos e palidez do disco óptico. Essa tríade, visível ao exame de fundo de olho, é o sinal de alerta que todo oftalmologista deve reconhecer imediatamente.
A prevalência da RP aumentou de 25,25 para 39,26 por 100.000 habitantes no período estudado por Abbass et al. no AJO 2025 — um aumento que reflete tanto o envelhecimento populacional quanto a melhora nos métodos diagnósticos, especialmente o sequenciamento genético de nova geração (NGS).
A genética da Retinose Pigmentar: mais de 90 genes, mais de 3.100 mutações
A complexidade genética da Retinose Pigmentar é extraordinária. Mais de 90 genes foram associados à doença, com mais de 3.100 mutações documentadas — e esse número cresce a cada ano com o avanço das técnicas de sequenciamento. Essa heterogeneidade genética explica por que duas pessoas com o mesmo diagnóstico clínico podem ter progressões completamente diferentes.
| Padrão de Herança | Frequência | Genes Principais | Características |
|---|---|---|---|
| Autossômica Dominante | 30–40% dos casos | RHO, RP1, PRPH2 | Início mais tardio, progressão mais lenta |
| Autossômica Recessiva | 50–60% dos casos | USH2A, PDE6A, PDE6B | Início mais precoce, progressão mais grave |
| Ligada ao X | 5–15% dos casos | RPGR, RP2 | Afeta principalmente homens; formas graves |
Aproximadamente 40% dos casos são esporádicos — sem história familiar conhecida — o que frequentemente atrasa o diagnóstico. A forma ligada ao X, embora menos frequente, tende a ser a mais grave, com início na infância e progressão rápida para cegueira legal.
Um dado relevante para o aconselhamento genético: a RP pode ocorrer de forma isolada (não sindrômica) ou como parte de síndromes sistêmicas. A Síndrome de Usher — que combina RP com surdez neurossensorial — é a síndrome mais comum associada à RP, causada principalmente por mutações no gene USH2A. Outras síndromes incluem Bardet-Biedl, Alport e, como detalhado em artigo separado neste blog, a Doença de Refsum.
Como a doença progride: da cegueira noturna à visão em túnel
A progressão da Retinose Pigmentar segue um padrão relativamente previsível, embora a velocidade varie enormemente entre indivíduos e formas genéticas.
Fase inicial (adolescência a 30 anos): O primeiro sintoma é quase sempre a nictalopia — dificuldade para enxergar em ambientes com pouca luz. O paciente relata dificuldade para dirigir à noite, tropeçar em objetos em ambientes mal iluminados e demorar mais para se adaptar ao escuro. Nessa fase, a visão central está preservada e o paciente frequentemente não percebe a gravidade da situação.
Fase intermediária (30 a 50 anos): A perda do campo visual periférico torna-se evidente. O paciente desenvolve a chamada visão em túnel — enxerga bem o que está diretamente à sua frente, mas não percebe objetos nas laterais. Acidentes de trânsito, quedas e colisões com objetos tornam-se frequentes. O eletrorretinograma (ERG) mostra amplitudes progressivamente reduzidas.
Fase avançada (após os 40 anos): A degeneração dos cones compromete a visão central. A leitura, o reconhecimento de faces e as atividades cotidianas tornam-se progressivamente impossíveis. Muitos pacientes atingem cegueira legal (acuidade visual <20/200 ou campo visual <20°) antes dos 40 anos, especialmente nas formas ligadas ao X e autossômicas recessivas.
Complicações adicionais incluem catarata subcapsular posterior (presente em até 50% dos pacientes) e edema macular cistoide (EMC) — ambos tratáveis e que, quando não diagnosticados, aceleram a perda visual.
Diagnóstico: o papel do ERG, OCT e do teste genético
O diagnóstico da Retinose Pigmentar é fundamentalmente clínico e eletrofisiológico, mas o teste genético tornou-se indispensável para o manejo moderno da doença.
O Eletrorretinograma (ERG) é o exame padrão-ouro. Mede a resposta elétrica da retina à estimulação luminosa e detecta a disfunção dos bastonetes e cones antes mesmo das alterações fundoscópicas. Na RP, o ERG mostra amplitudes reduzidas ou extintas, especialmente na resposta escotópica (bastonetes).
A Tomografia de Coerência Óptica (OCT) permite visualizar a estrutura da retina em cortes de alta resolução. Na RP, observa-se perda progressiva da zona elipsoide (camada dos fotorreceptores), afinamento da retina periférica e, nas fases avançadas, comprometimento da região macular. O OCT também é fundamental para detectar o edema macular cistoide.
O Campo Visual Computadorizado documenta a extensão da perda periférica e monitora a progressão ao longo do tempo. O padrão típico é um escotoma em anel que se expande progressivamente.
O Teste Genético por NGS (sequenciamento de nova geração) identifica a mutação causadora em 60–70% dos casos com painéis de distrofias retinianas. O diagnóstico genético é fundamental para confirmar o diagnóstico, determinar o padrão de herança, orientar o aconselhamento familiar e, cada vez mais, identificar pacientes elegíveis para terapias gênicas específicas.
Tratamentos: da vitamina A à terapia gênica
O único tratamento aprovado: Luxturna
Em dezembro de 2017, a FDA aprovou o Luxturna (voretigene neparvovec), desenvolvido pela Spark Therapeutics e comercializado internacionalmente pela Novartis — tornando-se a primeira terapia gênica aprovada para uma doença genética nos Estados Unidos e um marco histórico na oftalmologia.
O Luxturna é indicado para pacientes com mutações bialélicas no gene RPE65, que afeta aproximadamente 1.000 a 2.000 pessoas nos EUA. O tratamento consiste em uma injeção subretiniana de um vetor viral AAV2 carregando uma cópia funcional do gene RPE65. Os resultados clínicos demonstram melhora significativa da sensibilidade à luz, da mobilidade em ambientes com pouca iluminação e da qualidade de vida.
No Brasil, o Luxturna ainda não tem registro na ANVISA, mas pacientes com mutações em RPE65 podem buscar acesso por vias de importação excepcional. O Dr. Fernando Drudi e a Dra. Priscilla Almeida orientam pacientes sobre as opções disponíveis no contexto brasileiro.
Terapias gênicas em ensaios clínicos (2024–2026)
O pipeline de terapias gênicas para RP é um dos mais ativos da oftalmologia. Dados de 2026 identificam candidatos principais em ensaios clínicos avançados:
| Terapia | Empresa | Gene-alvo | Fase |
|---|---|---|---|
| Botaretigene sparoparvovec | J&J / MeiraGTx | RPGR (XLRP) | Fase III |
| Laruparetigene zovaparvovec | Beacon Therapeutics | RPGR (XLRP) | Fase II/III |
| MCO-010 | Nanoscope Therapeutics | Gene-agnóstico | Fase II/III |
| GS030 | Gensight Biologics | Gene-agnóstico | Fase I/II |
| OCU400 | Ocugen | NR2E3 (gene-agnóstico) | Fase II/III |
A revolução das terapias gene-agnósticas
O maior avanço conceitual no tratamento da RP nos últimos anos é o desenvolvimento de terapias gene-agnósticas — abordagens que funcionam independentemente da mutação genética específica do paciente. Isso é revolucionário porque significa que, em vez de precisar de um tratamento diferente para cada um dos 90+ genes causadores de RP, uma única terapia poderia beneficiar a maioria dos pacientes.
A optogenética é a abordagem mais promissora nesse grupo. O princípio é elegante: como os fotorreceptores (bastonetes e cones) estão degenerados, a terapia introduz genes de proteínas sensíveis à luz (opsinas) em outras células da retina que sobrevivem — como as células ganglionares ou bipolares — transformando-as em fotorreceptores funcionais. O MCO-010 (Nanoscope) e o GS030 (Gensight) são os candidatos mais avançados nessa categoria.
Outra abordagem gene-agnóstica inovadora é o OCU400 (Ocugen), que utiliza o gene NR2E3 — um fator de transcrição nuclear que regula a sobrevivência dos fotorreceptores — como uma espécie de "regulador mestre" capaz de proteger os fotorreceptores independentemente da mutação causadora da RP.
CRISPR/Cas9: editando o DNA da retina
A tecnologia CRISPR/Cas9 está avançando para ensaios clínicos em distrofias retinianas. Uma revisão publicada em 2025 (Geiger et al.) apresenta uma análise abrangente das tecnologias CRISPR sendo exploradas como intervenções terapêuticas para doenças retinianas hereditárias. A edição direta do gene defeituoso, em vez de simplesmente adicionar uma cópia funcional, representa o próximo passo na evolução das terapias gênicas.
Tratamentos de suporte: o que funciona hoje
Enquanto as terapias gênicas avançam, os tratamentos de suporte continuam sendo a base do manejo da RP na prática clínica. A vitamina A palmitato (15.000 UI/dia) demonstrou retardar a progressão da RP em aproximadamente 2 anos em estudos clínicos, embora seu uso deva ser monitorado por risco de toxicidade hepática e teratogenicidade. O DHA (ácido docosahexaenoico, ômega-3) pode potencializar o efeito da vitamina A em alguns subgrupos.
A proteção contra luz UV com óculos de filtro adequado é recomendada para reduzir o estresse oxidativo na retina. O tratamento das complicações — catarata subcapsular posterior com cirurgia e edema macular cistoide com inibidores da anidrase carbônica (acetazolamida) ou anti-VEGF — pode recuperar visão significativa em pacientes selecionados.
O impacto psicossocial: além da perda visual
A Retinose Pigmentar não afeta apenas os olhos. Pacientes com RP apresentam taxas significativamente elevadas de ansiedade, depressão e isolamento social. A incerteza sobre a progressão da doença, o impacto no emprego e na independência, e o estigma social associado à deficiência visual contribuem para uma carga psicossocial substancial.
O aconselhamento genético é fundamental não apenas para informar sobre o risco de transmissão aos filhos, mas também para preparar o paciente e a família para o curso da doença. A reabilitação visual — com auxílios ópticos, tecnologias assistivas e treinamento de orientação e mobilidade — deve ser iniciada precocemente, antes que a perda visual seja severa.
Quando procurar um especialista em retina
O diagnóstico precoce da Retinose Pigmentar é fundamental para maximizar as opções terapêuticas disponíveis — especialmente com o avanço das terapias gênicas, que tendem a ser mais eficazes quando iniciadas antes da perda completa dos fotorreceptores. Procure um especialista em retina se você ou um familiar apresentar dificuldade para enxergar à noite ou em ambientes com pouca luz, sensação de que o campo visual está se estreitando, dificuldade para enxergar objetos nas laterais (visão periférica), histórico familiar de cegueira ou problemas de visão de causa desconhecida, ou diagnóstico de surdez associado a problemas de visão (suspeita de Síndrome de Usher).
Na Drudi e Almeida Oftalmologia, o Instituto da Retina dispõe de tecnologia de última geração para o diagnóstico e acompanhamento da Retinose Pigmentar, incluindo ERG de última geração, OCT de alta resolução, campo visual computadorizado e painel genético de distrofias retinianas. O Dr. Fernando Drudi e a Dra. Priscilla Almeida orientam cada paciente sobre as melhores opções disponíveis, incluindo o acesso a ensaios clínicos de terapias gênicas quando aplicável.
A ciência avança. O diagnóstico precoce é o primeiro passo para aproveitar cada avanço.
Referências científicas
- Suleman N, et al. Current understanding on Retinitis Pigmentosa: a literature review. Front Ophthalmol. 2025;5:1600283. PMC12198980.
- Abbass NJ, et al. Trends and Disparities in the Incidence and Prevalence of Retinitis Pigmentosa. Am J Ophthalmol. 2025. doi:10.1016/j.ajo.2025.00375.
- Calderón RAM, et al. Retinitis Pigmentosa: From Genetic Insights to Innovative Therapeutic Approaches. PMC12298941. 2025.
- Da Palma MM, Salles MV, Sallum JMF. Terapia gênica para doenças da retina. Rev Bras Oftalmol. 2025. SciELO Brasil.
- Geiger AB, et al. Shining light on CRISPR/Cas9 therapeutics for inherited retinal diseases. Sci Direct. 2025. doi:10.1016/j.preteyeres.2025.000497.
- DelveInsight. 8 Emerging Gene Therapies for Retinitis Pigmentosa Treatment. March 2026.
Aviso importante: Este conteúdo tem caráter exclusivamente educativo e informativo. Não substitui a consulta, o diagnóstico ou o tratamento por um médico oftalmologista. Em caso de sintomas ou dúvidas sobre sua saúde ocular, procure atendimento médico especializado.
Guia Definitivo: Retina em São Paulo (2026)
Retinopatia diabética, DMRI, anti-VEGF e vitrectomia. Elaborado com 8 referências científicas de alto impacto.